-

Genome-wide association studies (GWAS) aim to identify associations of genotypes with phenotypes by testing for differences in the allele frequency of genetic variants between individuals. GWAS can consider copy-number variants or sequence variations in the human genome, although the most commonly studied genetic variants in GWAS are single-nucleotide polymorphisms (SNPs). GWAS typically report blocks of correlated SNPs that all show a statistically significant association with the trait of interest, known as genomic risk loci.
Conducting GWAS_selecting study population
•GWAS often require very large sample sizes to identify reproducible genome-wide significant associations and the desired sample size can be determined using power calculations in software tools such as CaTS or GPC. Study designs can involve the inclusion of cases and controls when the trait of interest is dichotomous, or quantitative measurements on the whole study sample when the trait is quantitative. In addition, one can choose between population-based and family-based designs.
•GWAS can be conducted using data from resources such as biobanks or cohorts. Even when new data have been collected inhouse, these will typically be co-analysed with data from pre-existing resources; collecting new data is usually required when more refined phenotyping is desired.•recruitment strategies must be carefully considered as these can induce collider bias and other forms of bias in the resultant data.
Ex) UK Biobank recruit participants through a volunteer-based strategy, which results in participants who are, on average, healthier, wealthier and more educated than the general population. Individual cohorts with detailed clinical measures may not be able to meet the required sample size; in these cases, ‘proxy’ phenotypes that are easier to measure. (ex. educational attainment -> intelligence)
Overview of steps for conducting GWAS
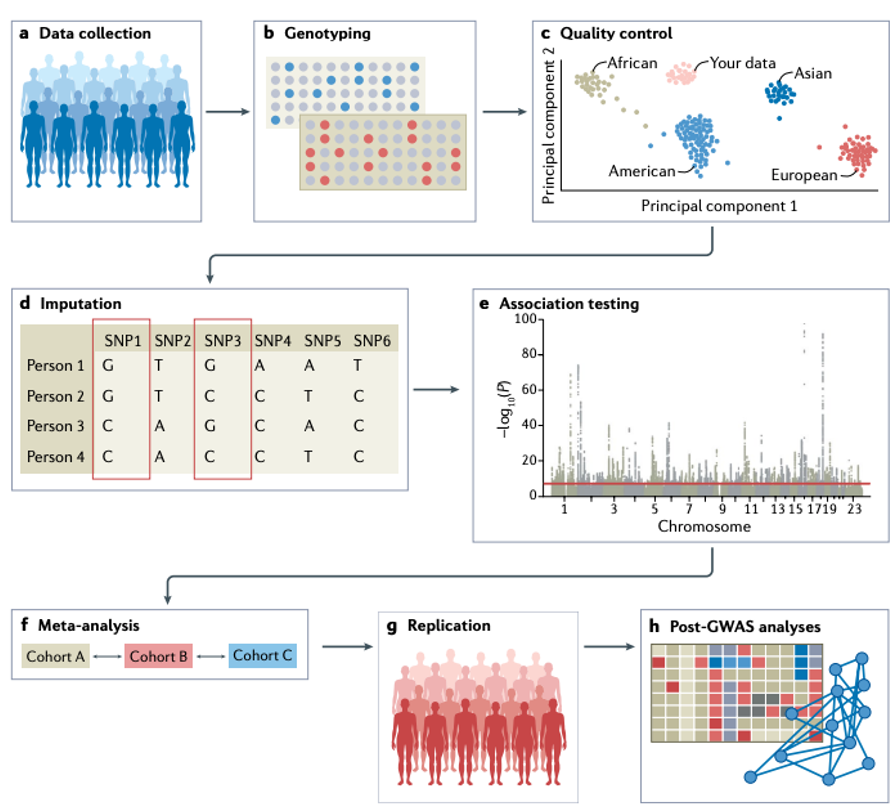
Genotyping
•Genotyping of individuals is typically done using microarrays for common variants or nextgeneration sequencing methods such as WES or WGS that also include rare variants. Microarray-based genotyping is the most commonly used method for obtaining genotypes for GWAS owing to the current cost of nextgeneration sequencing.
•However, the choice of genotyping platform depends on many factors and tends to be guided by the purpose of the GWAS.•Ideally, WGS — which determines nearly every genotype of a full genome — is preferred over WES and microarrays, and is expected to become the method of choice over the next couple of years with the increasing availability of low-cost WGS technologyData processing
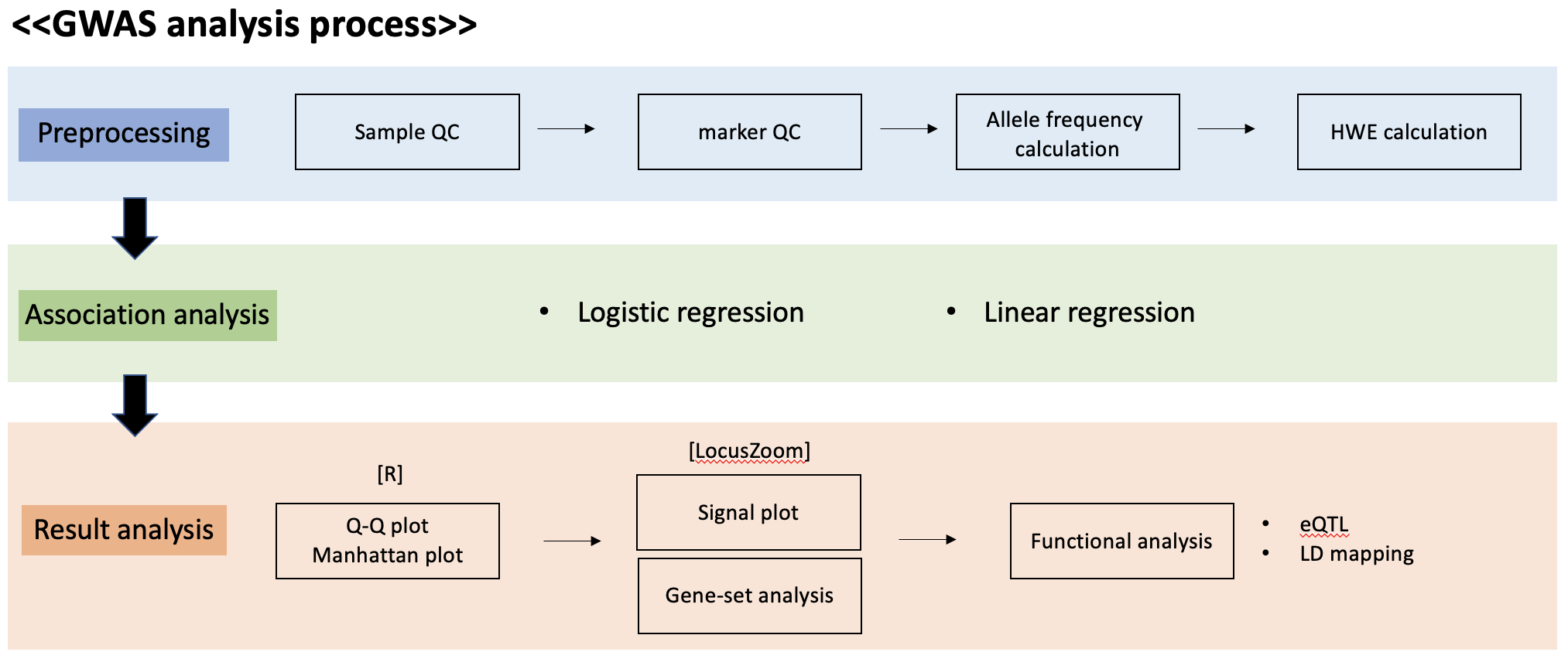
•Input files for a GWAS include anonymized individual ID numbers, coded family relations between individuals, sex, phenotype information, covariates, genotype calls for all called variants and information on the genotyping batch.•Generating reliable results from GWAS requires careful quality control, such as removing rare or monomorphic variants, removing variants that are not in Hardy–Weinberg equilibrium, filtering SNPs that are missing from a fraction of individuals in the cohort, identifying and removing genotyping errors, and ensuring that phenotypes are well matched with genetic data, often by comparing self-reported sex versus sex based on the X and Y chromosomes. (PLINK can be used for quality control steps)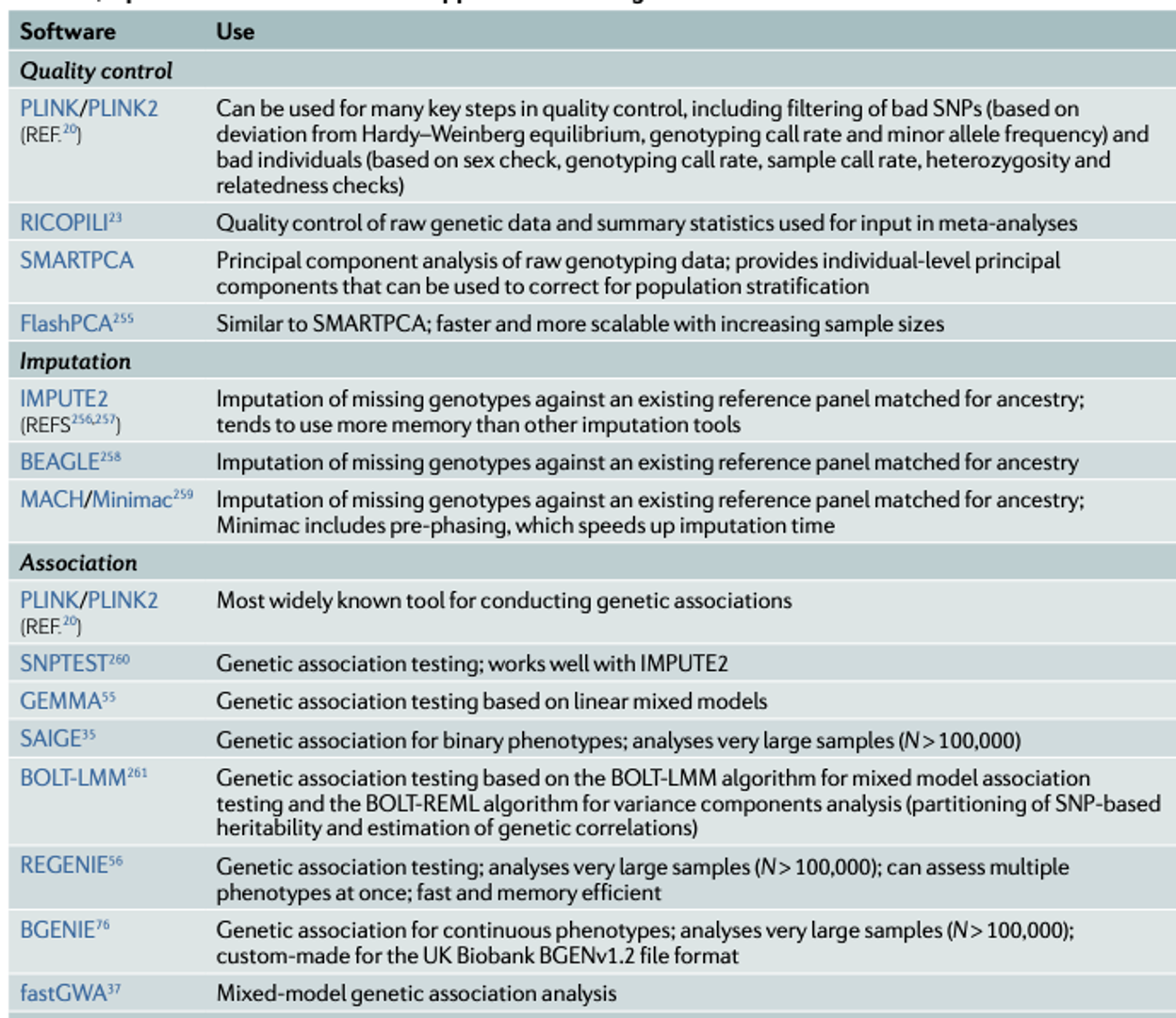
Imputation workflow
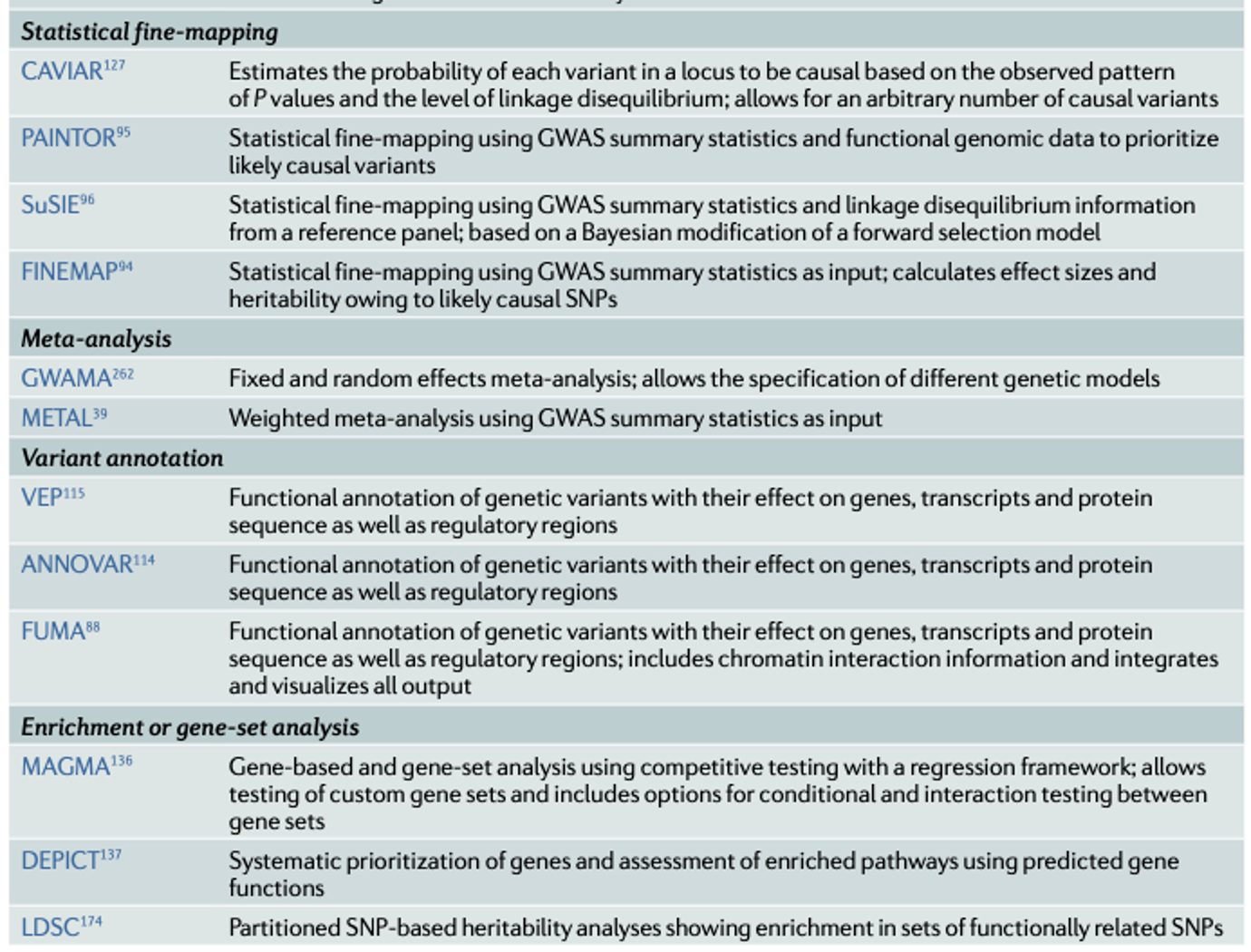
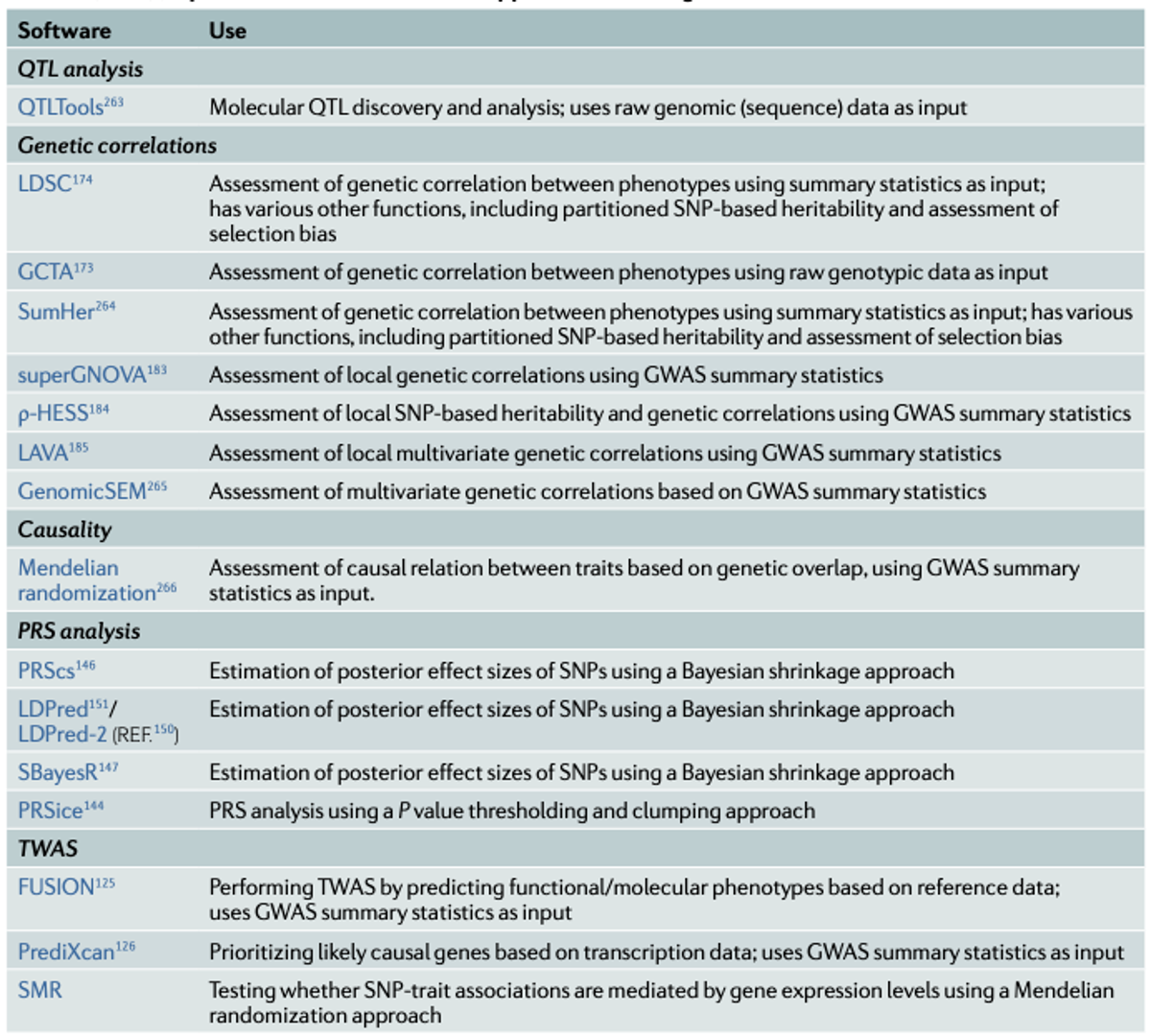
•Imputation of ungenotyped single-nucleotide polymorphisms (SNPs) can be done by using online imputation servers such as the Michigan Imputation Server or the TOPMed Imputation Server. Alternatively, one can carry out the imputation locally, using tools such as IMPUTE2, BEAGLE, MACH and SHAPEIT2. Imputation involves several steps.
•Statistically phase individual genotypes
•Decide whether to use hard calls or weight for uncertainty•Select an appropriate reference population panel•Convert reference panel and target population into the same genomic build•Check strand issues, resolve issues between different platforms, possibly remove ambiguous SNPs•Check for unusual minor allele frequencies and patterns of linkage disequilibrium between reference panel and target data•Impute missing genotypes against the selected population panel, ideally using cluster computing resources to distribute analysis jobs, or using an imputation server•Check imputation quality and possibly remove badly imputed SNPs (ex), those with an info score <0.7)
Testing for associations
•Typically in GWAS, linear or logistic regression models are used to test for associations, depending on whether the phenotype is continuous or binary. Covariates such as age, sex and ancestry are included to account for stratification and avoid confounding effects from demographic factors, with the caveat that this may reduce statistical power for binary traits in ascertained samples. Including an additional random effect term — which is individual-specific in linear or logistic mixed models to account for genetic relatedness among individuals — can improve statistical power for genomic discovery and increase control for stratification at the cost of requiring greater computational resources (although this limitation can be addressed by using tools such as fastGWA).•When conducting a GWAS, it should be noted that the genotypes of genetic variants that are physically close together are not independent as they tend to be in linkage disequilibrium; this dependency of tests should also be considered when conducting a GWAS.
Accounting for false discovery
•Testing millions of associations between individual genetic variants and a phenotype of interest requires a stringent multiple-testing threshold to avoid false positives. The International HapMap Project and other studies have shown that there are approximately 1million independent common genetic variants across the human genome on average, resulting in a Bonferroni testing threshold of P< 5×10–8 (representing a false discovery rate of 0.05/106 ).
•The appropriate threshold might vary depending on the population; for example, a more stringent threshold may be needed for populations with larger effective population sizes or if the minor allele frequency thresholds for inclusion in a GWAS are lowered as sample sizes increase, as low minor allele frequency variants are typically not in linkage disequilibrium with common variants and, therefore, add a greater multiple testing burden.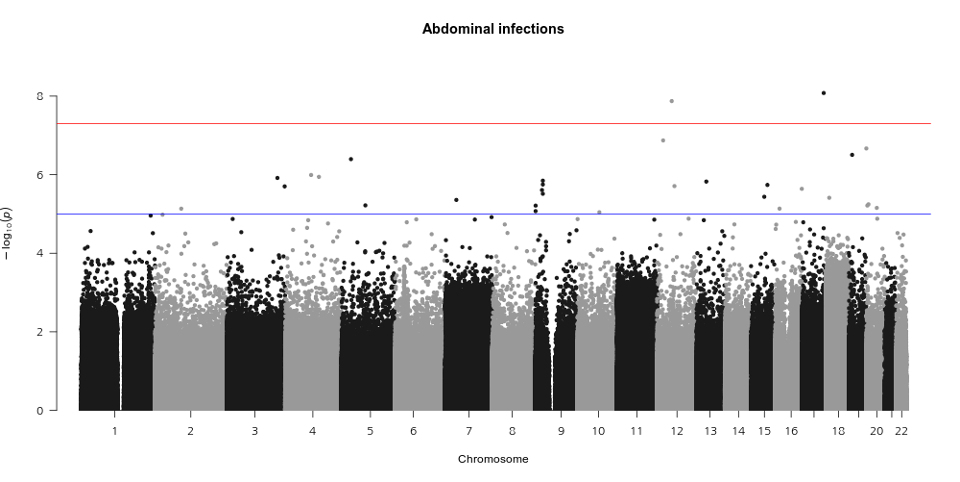
'BioinfoML' 카테고리의 다른 글
[ML] 기계학습 및 딥러닝 기초이론과 암 유전체 데이터 딥러닝 적용 실습 3강, 4강 (0) 2023.04.10 [ML] 기계학습 및 딥러닝 기초이론과 암 유전체 데이터 딥러닝 적용 실습 1강, 2강 (0) 2023.03.28